Участник @васьвась написал в Влияние показателей ВН и ИС на определении стадии ВИЧ+:
@gremlin , ты считаешь вирионы плазмы. Как насчет других жидкостей, резервуаров? В иммунограмме считают СД4-лимфоциты. Но клеток с этим рецептором разных хватает. Соответственно и ВИЧ живет в разных.
Я тоже об этом подумал перед тем, как считать вирионы и CD4-T-клетки плазмы. Но это все в итоге особо не повлияет на расчет и мою версию выше.
1.) По поводу иных инфицированных клеток. Другими, нежели инфицированные CD4-T-лимфоциты, клетками я сходу пренебрег. Ибо их доля в персистенции ВИЧ менее 1%. И влияние их мало. Поэтому объяснить их персистенцией скачок ВН до миллионных нагрузок не получится.
2.) По поводу других источников виремии нежели плазма (резервуаров ВИЧ). Да, действительно доля Т-лимфоцитов в лимфоидных органах составляет около 98% от всех циркулирующих в организме лимфоцитов, а доля Т-лимфоцитов, находящихся в каждый момент времени в плазме – около 2%.
Но для нашего расчета ВН это не играет никакой роли.
Допустим, возьмем для удобства и наглядности упрощенную модель «лимф. узел – плазма крови», которая состоит только из одного лимфатического узла и трубопровода плазмы. Пусть 98 инфицированных Т-лимфоцитов кампактно находятся в данный момент времени в лимф. узле, а 2 Т-лимфоцита находятся в кровотоке (в плазме). Пусть для удобства расчета и наглядности объем циркулирующей плазмы составляет 1 мл. И пусть каждый лимфоцит каждые 30 минут выпускает по 4 вириона, а средний срок их жизни соответствует реальному и равен 30 минутам.
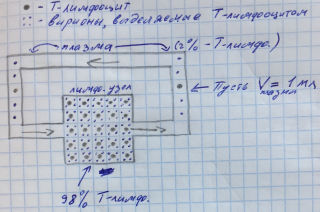
Таким образом, каждый лимфоцит в каждый период времени будут окружать по 4 выпущенных им вириона. Хоть в плазме (в более отдаленном друг от друга виде), хоть в лимфатическом узле (в более прижатом друг к другу виде). И при обмене (рециркуляции) Т-лимфоцитами между плазмой и лимф. узлом выходить из плазмы и входить в плазму каждый новый Т-лимфоцит все равно будет окруженный 4-я вирионами.
Допустим, у нас установилась такая скорость рециркуляции Т-лимфоцитов, при которой в плазму выходят и находятся там в каждый момент времени по 2 Т-лимфоцита. Выходят они из тесного лимф. узла в просторную плазму каждый с соответствующим соотношением вирионов и себя, т.е. с 4-мя вирионами. И получается, что в 1 мл плазмы у нас находятся 8 вирионов, а зная, количество продуцируемых клеткой в течение клиренса вирионов (4 вириона) мы можем поделить их на 4 и получить 2 Т-лимфоцита в мл. А если скорость рециркуляции уменьшается в 2 раза, то следовательно, вирионов вынесется в плазму пропорционально 1 Т-лимфоциту и ВН составит 4 копии/мл в нашем случае. И так далее.
Т.е., по итогу, резервуары инфицированных Т-лимфоцитов (лимф. узлы, например) на ВН в плазме влиять не будут.
P.S. единственное, что тут можно, конечно, представить, это то, что в резервуарах, допустим, вирионы не разрушаются за 30 минут (а в плазме разрушаются) и за счет этого происходит некое соответствующее этому накопление ВН и в плазме. Но, по сути, это будет та же моя версия выше с увеличенным клиренсом вирионов.