Вирусологические неудачи – устойчивость (резистентность) к терапии
-
Пользователь @аннаанна написал в Резистентность на ламивудин, выбор схемы, нужна помощь:
При таких данных, какая схема была бы оптимальной?
Тивикай+дарунавир 800+ритонавир. И пусть больше врач не мудрит. Уже намудрил.
-
Пользователь @васьвась написал в Резистентность на ламивудин, выбор схемы, нужна помощь:
Уже намудрил.
Замена калетры на интеленс,
То есть схема с Элпидой нагрузку не снижала, но он и дальше продолжил назначать ННИОТ.
-
Подайте уже кто-нибудь с яйцами заявление на этих идиотов-мудаков в белых халатах, пусть посидят за причинение тяжкого. В прокуратуре с ск палке/звездочке только рады будут. Остальным мб наука будет, иначе не поумнеют.
-
@васьвась спасибо!
-
@аннаанна вариантов тут больше одного, потому, лучше бы не расшифровку, а коды мутаций. По расшифровке особо советов не дать, или они будут весьма общего толка.
-
@ilya-antipin Здравствуйте, Илья Игоревич! При мне медсестра принесла анализ, был лист только с этими данными. На следующей неделе к врачу, обязательно спрошу, но вопрос в том, что скорее всего будут менять терапию, а я уже не уверена в верности назначений препаратов. Сама в теме полностью еще не разобралась.
-
Пользователь @аннаанна написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
а я уже не уверена в верности назначений препаратов
У вас не работает один препарат в схеме, да, такую схему пить нельзя.
При только таких данных один из лучших вариантов рабочей схемы Васьвась написал
-
@stuppy и тот, на момент теста, сегодня – уже бабка на двое сказала. Там бы по уму отменять безопаснее было бы, до результата.
-
Отличная возможность для наивных пациентов МСМ из Москвы
📌📌📌Федеральный научно-методический Центр по профилактике и борьбе со СПИДом( Космодамианская набережная, д.22, стр.1а)
проводит набор МСМ на клиническое исследование по ЛУ (лекарственной устойчивости или иначе резистентности к арвт).
Требование - берут только тех, кто никогда не пил терапию. Врать не стоит, это будет проверяться.
Обращаться к Поповой Анне Анатольевне.
С собой нужно будет иметь результат ИС (CD4). -
Извините,может не туда написала
-
@аннаанна извиняюсь, может не к месту, хотела о своём опыте вам написать : пила несколько лет тивикай/абакавир/ламивудин, всегда вн н/о, при том, что ламивудин у меня абсолютно резистентный, и абакавир, возможно с неск-кими минорными мутациями. И на дарунавир 800/ритонавир с теми же препарами - тоже вн всегда н/о. Я к тому веду, что судя по рекомендациям, мне тоже было положено наверное выдать более усиленную схему, но тем не менее, выше две упомянутые схемы работали без всплесков вн, пила их обе по несколько лет, также и сейчас продолжаю принимать
-
@monaliza Вот мне сегодня выписала такую же схему: ламивудин, абакавир, дарунавир и ритонавир. Ранее была на элпиде, ламивудине и абакавире, но вирусная не становилась неопределяемой. И никакого теста на резистентность не делали.
-
@monaliza
спасибо, что поделились опытом. назначили тивикай тенофовир ламивудин. Зная, что у вас схема работает схема с ламивудином с резистентностью к нему, есть надежда, что время не будет потрачено снова впустую. В усиленной схеме, как рекомендовали выше, отказали. -
Неудача схем на основе ингибиторов интегразы может быть связана с новой формой резистентности
Иногда Джулука или даже Эвиплера может стать лучшим выбором, значит.
-
Пользователь @васьвась написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
Неудача схем на основе ингибиторов интегразы может быть связана с новой формой резистентности
Иногда Джулука или даже Эвиплера может стать лучшим выбором, значит.
@Ilya-Antipin , этот материал напомнил о проблеме иммунологических неудач. Когда в плазме нагрузки нет, а ИС не растет или даже снижается.
Похоже в этом задействован такой Env-опосредованный механизм передачи вируса от клетки к клетке.
Вопрос - чем его блокировать.
@Gremlin , что думаешь?Вот еще кое-что копнул, для подробностей:
-
Пользователь @васьвась написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
@Ilya-Antipin , этот материал напомнил о проблеме иммунологических неудач. Когда в плазме нагрузки нет, а ИС не растет или даже снижается.
Похоже в этом задействован такой Env-опосредованный механизм передачи вируса от клетки к клетке.
Вопрос - чем его блокировать.
@Gremlin , что думаешь?По этому материалу, лично у меня, возникает масса вопросов и не так уж много внятных ответов:
1.) Если такой способ передачи ВИЧ-инфекции от клетки к клетке действительно так эффективен
This was shown to be the case when the researchers cultivated HIV-infected and uninfected cells together: after 24 hours, three times more cells were newly infected if the infected cells contained HIV with the Env7 mutations than with none.
, то тогда и без давления АРВТ у всех хронически инфицированных пациентов, по идее, с течением времени должны отбираться в основном эти 7Env-мутанты и тогда клеток, инфицированных такими мутантами, должно быть большинство.
2.)
Похоже в этом задействован такой Env-опосредованный механизм передачи вируса от клетки к клетке.
Если бы такой способ убивал CD4+ клетки, несмотря на присутствие ингибиторов интегразы и обратной транскриптазы, то как объяснить те же данные, которые, помнится, приводил где-то здесь Илья Игоревич о преимуществе в плане роста ИС схем с ингибиторами интегразы над схемами с ИП и ННИОТами. А также, учитывая результаты данного исследования о разнице в чувствительности этих 7Env-мутантов к ИИ и к Н(Н)ИОТАМ
It was found that the ‘Env7’ mutations cut HIV’s sensitivity to dolutegravir and cabotegravir by a factor of 2000 and sensitivity to raltegravir by 400. Other drug classes were affected by these mutations too, but not nearly as much: the Env7 mutations cut sensitivity to the reverse transcriptase inhibitors emtricitabine and efavirenz elevenfold and to rilpivirine fourfold, and to the protease inhibitor darunavir sixfold.
, можно предположить, что Н(Н)ИОТы должны гораздо лучше справляться с «множественным инфицированием» (MOI)
This process introduces a larger ‘dose’ of the virus into the infected cells than infection via virions. Integrase inhibitors deal less well with this greater so-called ‘multiplicity of infection’ (MOI) than other classes.
клеток этими «шустрыми» Env-мутантами на более ранней стадии жизненного цикла ВИЧ (а именно на стадии обратной транскрипции), чем перенос уже обратнооттранскрибированной цепи вирусной ДНК интегразой и таким образом препятствовать распространению ВИЧ в другие клетки и, как следствие, останавливать смерть клеток.
Или же уменьшения в несколько раз эффективности НИОТов достаточно для прорыва Env-мутантов…?3.) Помнится также я как-то писал материал, где из исследований выходило, что основным источником смертей CD4+ клеток является пироптоз, вызванный в основном накоплением в цитоплазме клеток неполных обратных транскриптов. То есть для падения ИС вовсе необязательно доходить до стадии интеграции вирусной ДНК в геном хозяина. То есть для падения ИС нет большого выигрыша от успешности массированного набега Env-мутантов в клетку и их победой над ингибиторами интегразы.
4.) Согласно данному исследованию эффективность межклеточного инфицирования клеток Env-мутантами в 3 раза выше, чем при межклеточном инфицировании диким вирусом
This was shown to be the case when the researchers cultivated HIV-infected and uninfected cells together: after 24 hours, three times more cells were newly infected if the infected cells contained HIV with the Env7 mutations than with none.
в отличие от меньшей эффективности при инфицировании свободными вирионами
In cells infected by unmutated free-floating viruses, the levels of HIV’s reverse transcriptase (RT) enzyme – a measure of viral replication – peaked at week 8 after infection, but at week 12 in viruses with the seven envelope mutations.
При этом чувствительность Env-мутантов к DTG снижена в 2000 раз, к RAL в 400 раз, к FTC и EFV в 11 раз, к RPV в 4 раза, к DRV в 6 раз. И авторы делают предположение, что причиной, по которой может происходить такая значительная потеря чувствительности к ингибиторам интегразы может быть массированность атаки кучи «шустрых» Env-мутантов при межклеточном инфицировании
The researchers theorised that the envelope mutations enhanced HIV’s ability to cross from one cell to another by direct transfer, rather than with free-floating virions.
и то, что молекулы ингибиторов интегразы в клетке попросту не успевают связать такую кучу интеграз там.
Cell-to-cell transfer may happen less often than virion transfer but, when it does, it may introduce thirty times as much viral protein into the cell than viral particles. The reason INSTIs cope less well with what’s called the greater multiplicity of infectivity (MoI) in cell-to-cell infection is that they simply can’t ‘get at’ the integrase strand transfer site every time – they are overwhelmed by the higher MoI.
И тут тогда у меня возникает несколько вопросов:
а) откуда такая разница между чувствительностью Env-мутантов к Н(Н)ИОТам и к ингибиторам интегразы (ИИ) при множественной инфекции? Ведь то, что количество интеграз на вирион 50 – 100 штук, а количество обратных транскриптаз около 50 штук не говорит о том, что ингибиторов интегразы требуется в 2 раза больше. Так как количество цепей РНК в вирионе всего 2 и что обратная транскриптаза, что комплекс из 4-х интеграз (тетрамер)
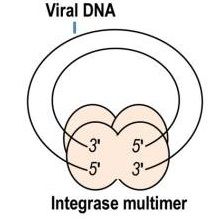
работают одновременно лишь с одной цепью вирусной РНК/ДНК в вирионе, а Н(Н)ИОТы и ИИ включаются именно в состав комплекса вРНК-ОТ и комплекса вДНК-тетрамер интеграз (интасома), а не работают с одиночными ОТ/интегразами в вирионе, то есть, по идее на один вирион требуется сопоставимое число Н(Н)ИОТов и ИИ и, следовательно, и Н(Н)ИОТы и ИИ должны одинаково успешно/безуспешно бороться с множественной инфекцией клетки «шустрыми» Env-мутантами;
б) также странно при такой теории множественной инфекции (MOI), что чувствительность Env-мутантов к RAL выше, чем к DTG, ведь сродство (прочность сцепления) DTG к интасоме ВИЧ выше, чем сродство RAL к ней, а также период полураспада комплекса интасома ВИЧ-DTG длиннее, чем комплекса интасома ВИЧ-RAL, то есть, по идее, DTG должен успешней противостоять массированной атаке Env-мутантов на клетку, чем RAL.
Или же внутриклеточные концентрации молекул RAL и Н(Н)ИОТов значительно выше, чем DTG и это позволяет им успешней противостоять множественной инфекции…?5.) Также возникает вопрос, почему при такой теории межклеточной множественной инфекции Env-мутантами чувствительность к ингибитору протеазы (ИП) DRV снижена у этих мутантов в 6 раз. Ведь, по идее, ингибиторам протеазы вообще фиолетово насколько много вирионов вошло в клетку. Они (ИП) работают на стадии созревания вириона после транскрипции из ДНК хозяина, не давая созреть вирионам. То есть, пусть, в клетку при межклеточном инфицировании вошло в 30 раз больше по сравнению с дикими вирионами Env-мутантных вирионов и какая-то часть из них пробилась в геном хозяина, тогда при активации данной клетки и транскрипции ее ДНК в созревших вирионах ИП блокируют протеазу ВИЧ и им (ИП) без разницы сколько до этого вошло вирионов в клетку, чтобы встроить свой геном в геном человека.
Или из-за большого количества атаковавших клетку вирионов в геном встроилось достаточно много вирусных ДНК и при транскрипции ДНК хозяина они все резко начали «выплевывать» много вирионов и на всех их (на их протеазы) стало не хватать базовой концентрации ИП в клетке…?Вопрос - чем его блокировать.
Теоретически, комбинация Н(Н)ИОТов с ИП должна лучше всего препятствовать межклеточной передаче и накоплению обратных транскриптов, тем самым, сохраняя CD4+. Но, тогда непонятно почему Н(Н)ИОТы в комбинации с ИИ лучше всех способствуют росту ИС…
-
Пользователь @gremlin написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
Пользователь @васьвась написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
@Ilya-Antipin , этот материал напомнил о проблеме иммунологических неудач. Когда в плазме нагрузки нет, а ИС не растет или даже снижается.
Похоже в этом задействован такой Env-опосредованный механизм передачи вируса от клетки к клетке.
Вопрос - чем его блокировать.
@Gremlin , что думаешь?По этому материалу, лично у меня, возникает масса вопросов и не так уж много внятных ответов:
1.) Если такой способ передачи ВИЧ-инфекции от клетки к клетке действительно так эффективен
This was shown to be the case when the researchers cultivated HIV-infected and uninfected cells together: after 24 hours, three times more cells were newly infected if the infected cells contained HIV with the Env7 mutations than with none.
, то тогда и без давления АРВТ у всех хронически инфицированных пациентов, по идее, с течением времени должны отбираться в основном эти 7Env-мутанты и тогда клеток, инфицированных такими мутантами, должно быть большинство.
2.)
Похоже в этом задействован такой Env-опосредованный механизм передачи вируса от клетки к клетке.
Если бы такой способ убивал CD4+ клетки, несмотря на присутствие ингибиторов интегразы и обратной транскриптазы, то как объяснить те же данные, которые, помнится, приводил где-то здесь Илья Игоревич о преимуществе в плане роста ИС схем с ингибиторами интегразы над схемами с ИП и ННИОТами. А также, учитывая результаты данного исследования о разнице в чувствительности этих 7Env-мутантов к ИИ и к Н(Н)ИОТАМ
It was found that the ‘Env7’ mutations cut HIV’s sensitivity to dolutegravir and cabotegravir by a factor of 2000 and sensitivity to raltegravir by 400. Other drug classes were affected by these mutations too, but not nearly as much: the Env7 mutations cut sensitivity to the reverse transcriptase inhibitors emtricitabine and efavirenz elevenfold and to rilpivirine fourfold, and to the protease inhibitor darunavir sixfold.
, можно предположить, что Н(Н)ИОТы должны гораздо лучше справляться с «множественным инфицированием» (MOI)
This process introduces a larger ‘dose’ of the virus into the infected cells than infection via virions. Integrase inhibitors deal less well with this greater so-called ‘multiplicity of infection’ (MOI) than other classes.
клеток этими «шустрыми» Env-мутантами на более ранней стадии жизненного цикла ВИЧ (а именно на стадии обратной транскрипции), чем перенос уже обратнооттранскрибированной цепи вирусной ДНК интегразой и таким образом препятствовать распространению ВИЧ в другие клетки и, как следствие, останавливать смерть клеток.
Или же уменьшения в несколько раз эффективности НИОТов достаточно для прорыва Env-мутантов…?3.) Помнится также я как-то писал материал, где из исследований выходило, что основным источником смертей CD4+ клеток является пироптоз, вызванный в основном накоплением в цитоплазме клеток неполных обратных транскриптов. То есть для падения ИС вовсе необязательно доходить до стадии интеграции вирусной ДНК в геном хозяина. То есть для падения ИС нет большого выигрыша от успешности массированного набега Env-мутантов в клетку и их победой над ингибиторами интегразы.
4.) Согласно данному исследованию эффективность межклеточного инфицирования клеток Env-мутантами в 3 раза выше, чем при межклеточном инфицировании диким вирусом
This was shown to be the case when the researchers cultivated HIV-infected and uninfected cells together: after 24 hours, three times more cells were newly infected if the infected cells contained HIV with the Env7 mutations than with none.
в отличие от меньшей эффективности при инфицировании свободными вирионами
In cells infected by unmutated free-floating viruses, the levels of HIV’s reverse transcriptase (RT) enzyme – a measure of viral replication – peaked at week 8 after infection, but at week 12 in viruses with the seven envelope mutations.
При этом чувствительность Env-мутантов к DTG снижена в 2000 раз, к RAL в 400 раз, к FTC и EFV в 11 раз, к RPV в 4 раза, к DRV в 6 раз. И авторы делают предположение, что причиной, по которой может происходить такая значительная потеря чувствительности к ингибиторам интегразы может быть массированность атаки кучи «шустрых» Env-мутантов при межклеточном инфицировании
The researchers theorised that the envelope mutations enhanced HIV’s ability to cross from one cell to another by direct transfer, rather than with free-floating virions.
и то, что молекулы ингибиторов интегразы в клетке попросту не успевают связать такую кучу интеграз там.
Cell-to-cell transfer may happen less often than virion transfer but, when it does, it may introduce thirty times as much viral protein into the cell than viral particles. The reason INSTIs cope less well with what’s called the greater multiplicity of infectivity (MoI) in cell-to-cell infection is that they simply can’t ‘get at’ the integrase strand transfer site every time – they are overwhelmed by the higher MoI.
И тут тогда у меня возникает несколько вопросов:
а) откуда такая разница между чувствительностью Env-мутантов к Н(Н)ИОТам и к ингибиторам интегразы (ИИ) при множественной инфекции? Ведь то, что количество интеграз на вирион 50 – 100 штук, а количество обратных транскриптаз около 50 штук не говорит о том, что ингибиторов интегразы требуется в 2 раза больше. Так как количество цепей РНК в вирионе всего 2 и что обратная транскриптаза, что комплекс из 4-х интеграз (тетрамер)
работают одновременно лишь с одной цепью вирусной РНК/ДНК в вирионе, а Н(Н)ИОТы и ИИ включаются именно в состав комплекса вРНК-ОТ и комплекса вДНК-тетрамер интеграз (интасома), а не работают с одиночными ОТ/интегразами в вирионе, то есть, по идее на один вирион требуется сопоставимое число Н(Н)ИОТов и ИИ и, следовательно, и Н(Н)ИОТы и ИИ должны одинаково успешно/безуспешно бороться с множественной инфекцией клетки «шустрыми» Env-мутантами;
б) также странно при такой теории множественной инфекции (MOI), что чувствительность Env-мутантов к RAL выше, чем к DTG, ведь сродство (прочность сцепления) DTG к интасоме ВИЧ выше, чем сродство RAL к ней, а также период полураспада комплекса интасома ВИЧ-DTG длиннее, чем комплекса интасома ВИЧ-RAL, то есть, по идее, DTG должен успешней противостоять массированной атаке Env-мутантов на клетку, чем RAL.
Или же внутриклеточные концентрации молекул RAL и Н(Н)ИОТов значительно выше, чем DTG и это позволяет им успешней противостоять множественной инфекции…?5.) Также возникает вопрос, почему при такой теории межклеточной множественной инфекции Env-мутантами чувствительность к ингибитору протеазы (ИП) DRV снижена у этих мутантов в 6 раз. Ведь, по идее, ингибиторам протеазы вообще фиолетово насколько много вирионов вошло в клетку. Они (ИП) работают на стадии созревания вириона после транскрипции из ДНК хозяина, не давая созреть вирионам. То есть, пусть, в клетку при межклеточном инфицировании вошло в 30 раз больше по сравнению с дикими вирионами Env-мутантных вирионов и какая-то часть из них пробилась в геном хозяина, тогда при активации данной клетки и транскрипции ее ДНК в созревших вирионах ИП блокируют протеазу ВИЧ и им (ИП) без разницы сколько до этого вошло вирионов в клетку, чтобы встроить свой геном в геном человека.
Или из-за большого количества атаковавших клетку вирионов в геном встроилось достаточно много вирусных ДНК и при транскрипции ДНК хозяина они все резко начали «выплевывать» много вирионов и на всех их (на их протеазы) стало не хватать базовой концентрации ИП в клетке…?Вопрос - чем его блокировать.
Теоретически, комбинация Н(Н)ИОТов с ИП должна лучше всего препятствовать межклеточной передаче и накоплению обратных транскриптов, тем самым, сохраняя CD4+. Но, тогда непонятно почему Н(Н)ИОТы в комбинации с ИИ лучше всех способствуют росту ИС…
Короче говоря схема -дол рилпивирир получше чем дол ламивудин ?
-
Пользователь @basky написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
Короче говоря
чтобы было короче, нужно при обращении нажимать на кнопку “ответить”, а цитируют лишь определённые моменты, которые были непонятны и которые хотелось бы уточнить.
-
@sven понято
-
Пользователь @basky написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
Пользователь @gremlin написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
Пользователь @васьвась написал в Вирусологические неудачи – устойчивость (резистентность) к терапии:
@Ilya-Antipin , этот материал напомнил о проблеме иммунологических неудач. Когда в плазме нагрузки нет, а ИС не растет или даже снижается.
Похоже в этом задействован такой Env-опосредованный механизм передачи вируса от клетки к клетке.
Вопрос - чем его блокировать.
@Gremlin , что думаешь?По этому материалу, лично у меня, возникает масса вопросов и не так уж много внятных ответов:
1.) Если такой способ передачи ВИЧ-инфекции от клетки к клетке действительно так эффективен
This was shown to be the case when the researchers cultivated HIV-infected and uninfected cells together: after 24 hours, three times more cells were newly infected if the infected cells contained HIV with the Env7 mutations than with none.
, то тогда и без давления АРВТ у всех хронически инфицированных пациентов, по идее, с течением времени должны отбираться в основном эти 7Env-мутанты и тогда клеток, инфицированных такими мутантами, должно быть большинство.
2.)
Похоже в этом задействован такой Env-опосредованный механизм передачи вируса от клетки к клетке.
Если бы такой способ убивал CD4+ клетки, несмотря на присутствие ингибиторов интегразы и обратной транскриптазы, то как объяснить те же данные, которые, помнится, приводил где-то здесь Илья Игоревич о преимуществе в плане роста ИС схем с ингибиторами интегразы над схемами с ИП и ННИОТами. А также, учитывая результаты данного исследования о разнице в чувствительности этих 7Env-мутантов к ИИ и к Н(Н)ИОТАМ
It was found that the ‘Env7’ mutations cut HIV’s sensitivity to dolutegravir and cabotegravir by a factor of 2000 and sensitivity to raltegravir by 400. Other drug classes were affected by these mutations too, but not nearly as much: the Env7 mutations cut sensitivity to the reverse transcriptase inhibitors emtricitabine and efavirenz elevenfold and to rilpivirine fourfold, and to the protease inhibitor darunavir sixfold.
, можно предположить, что Н(Н)ИОТы должны гораздо лучше справляться с «множественным инфицированием» (MOI)
This process introduces a larger ‘dose’ of the virus into the infected cells than infection via virions. Integrase inhibitors deal less well with this greater so-called ‘multiplicity of infection’ (MOI) than other classes.
клеток этими «шустрыми» Env-мутантами на более ранней стадии жизненного цикла ВИЧ (а именно на стадии обратной транскрипции), чем перенос уже обратнооттранскрибированной цепи вирусной ДНК интегразой и таким образом препятствовать распространению ВИЧ в другие клетки и, как следствие, останавливать смерть клеток.
Или же уменьшения в несколько раз эффективности НИОТов достаточно для прорыва Env-мутантов…?3.) Помнится также я как-то писал материал, где из исследований выходило, что основным источником смертей CD4+ клеток является пироптоз, вызванный в основном накоплением в цитоплазме клеток неполных обратных транскриптов. То есть для падения ИС вовсе необязательно доходить до стадии интеграции вирусной ДНК в геном хозяина. То есть для падения ИС нет большого выигрыша от успешности массированного набега Env-мутантов в клетку и их победой над ингибиторами интегразы.
4.) Согласно данному исследованию эффективность межклеточного инфицирования клеток Env-мутантами в 3 раза выше, чем при межклеточном инфицировании диким вирусом
This was shown to be the case when the researchers cultivated HIV-infected and uninfected cells together: after 24 hours, three times more cells were newly infected if the infected cells contained HIV with the Env7 mutations than with none.
в отличие от меньшей эффективности при инфицировании свободными вирионами
In cells infected by unmutated free-floating viruses, the levels of HIV’s reverse transcriptase (RT) enzyme – a measure of viral replication – peaked at week 8 after infection, but at week 12 in viruses with the seven envelope mutations.
При этом чувствительность Env-мутантов к DTG снижена в 2000 раз, к RAL в 400 раз, к FTC и EFV в 11 раз, к RPV в 4 раза, к DRV в 6 раз. И авторы делают предположение, что причиной, по которой может происходить такая значительная потеря чувствительности к ингибиторам интегразы может быть массированность атаки кучи «шустрых» Env-мутантов при межклеточном инфицировании
The researchers theorised that the envelope mutations enhanced HIV’s ability to cross from one cell to another by direct transfer, rather than with free-floating virions.
и то, что молекулы ингибиторов интегразы в клетке попросту не успевают связать такую кучу интеграз там.
Cell-to-cell transfer may happen less often than virion transfer but, when it does, it may introduce thirty times as much viral protein into the cell than viral particles. The reason INSTIs cope less well with what’s called the greater multiplicity of infectivity (MoI) in cell-to-cell infection is that they simply can’t ‘get at’ the integrase strand transfer site every time – they are overwhelmed by the higher MoI.
И тут тогда у меня возникает несколько вопросов:
а) откуда такая разница между чувствительностью Env-мутантов к Н(Н)ИОТам и к ингибиторам интегразы (ИИ) при множественной инфекции? Ведь то, что количество интеграз на вирион 50 – 100 штук, а количество обратных транскриптаз около 50 штук не говорит о том, что ингибиторов интегразы требуется в 2 раза больше. Так как количество цепей РНК в вирионе всего 2 и что обратная транскриптаза, что комплекс из 4-х интеграз (тетрамер)
работают одновременно лишь с одной цепью вирусной РНК/ДНК в вирионе, а Н(Н)ИОТы и ИИ включаются именно в состав комплекса вРНК-ОТ и комплекса вДНК-тетрамер интеграз (интасома), а не работают с одиночными ОТ/интегразами в вирионе, то есть, по идее на один вирион требуется сопоставимое число Н(Н)ИОТов и ИИ и, следовательно, и Н(Н)ИОТы и ИИ должны одинаково успешно/безуспешно бороться с множественной инфекцией клетки «шустрыми» Env-мутантами;
б) также странно при такой теории множественной инфекции (MOI), что чувствительность Env-мутантов к RAL выше, чем к DTG, ведь сродство (прочность сцепления) DTG к интасоме ВИЧ выше, чем сродство RAL к ней, а также период полураспада комплекса интасома ВИЧ-DTG длиннее, чем комплекса интасома ВИЧ-RAL, то есть, по идее, DTG должен успешней противостоять массированной атаке Env-мутантов на клетку, чем RAL.
Или же внутриклеточные концентрации молекул RAL и Н(Н)ИОТов значительно выше, чем DTG и это позволяет им успешней противостоять множественной инфекции…?5.) Также возникает вопрос, почему при такой теории межклеточной множественной инфекции Env-мутантами чувствительность к ингибитору протеазы (ИП) DRV снижена у этих мутантов в 6 раз. Ведь, по идее, ингибиторам протеазы вообще фиолетово насколько много вирионов вошло в клетку. Они (ИП) работают на стадии созревания вириона после транскрипции из ДНК хозяина, не давая созреть вирионам. То есть, пусть, в клетку при межклеточном инфицировании вошло в 30 раз больше по сравнению с дикими вирионами Env-мутантных вирионов и какая-то часть из них пробилась в геном хозяина, тогда при активации данной клетки и транскрипции ее ДНК в созревших вирионах ИП блокируют протеазу ВИЧ и им (ИП) без разницы сколько до этого вошло вирионов в клетку, чтобы встроить свой геном в геном человека.
Или из-за большого количества атаковавших клетку вирионов в геном встроилось достаточно много вирусных ДНК и при транскрипции ДНК хозяина они все резко начали «выплевывать» много вирионов и на всех их (на их протеазы) стало не хватать базовой концентрации ИП в клетке…?Вопрос - чем его блокировать.
Теоретически, комбинация Н(Н)ИОТов с ИП должна лучше всего препятствовать межклеточной передаче и накоплению обратных транскриптов, тем самым, сохраняя CD4+. Но, тогда непонятно почему Н(Н)ИОТы в комбинации с ИИ лучше всех способствуют росту ИС…
Короче говоря схема -дол рилпивирир получше чем дол ламивудин ?
Короче говоря дол +рилпивирин это для резистентников нормал ,я так понял ?